|
Nach Angaben des Deutschen Kinderkrebsregisters repräsentieren Hirntumoren ca. 20% aller Tumoren im Kindes- und Jugendalter und sind damit die häufigsten soliden Tumoren in dieser Altersgruppe. Häufiger treten lediglich die Leukämien und anderen Erkrankungen des blutbildenden Systems auf. Man geht von ca. 300 ? 350 Hirntumor-Neuerkrankungen pro Jahr in Deutschland aus.
Die häufigsten Tumoren im Kindesalter sind Astrozytome (ca. 40%), Medulloblastome (ca. 20%) und Ependymome (ca. 10%). Weitere (eher seltene) Tumoren sind PNET (Primitive Neuroektodermale Tumoren), Kraniopharyngeome, Gangliogliome, Plexustumoren, Pinealistumoren, Keimzelltumoren und Meningeome.
Die häufigsten Symptome einer Hirntumor-Erkrankung im Kindesalter sind Kopfschmerzen, Übelkeit und Brechreiz, Gleichgewichtsstörungen und Schwindel, Störungen der Augenbewegung sowie Müdigkeit und vermehrtes Schlafbedürfnis. Seltener oder auch später im Krankheitsverlauf können Koordinationsstörungen, Lähmungen, Beeinträchtigung der Sehschärfe, Schwerhörigkeit oder ein Schiefhals (Torticollis) auftreten.
Über die Ursachen der Entstehung von Hirntumoren im Kindesalter ist wenig bekannt. Es gibt keine sicher nachgewiesenen Risikofaktoren, jedoch einige prädisponierende Erkrankungen, d.h., vorbestehende, meist bekannte Erkrankungen, die das Risiko einen Hirntumor zu entwickeln potentiell erhöhen. Dazu gehören unter anderem: Neurofibromatose, Tuberöse Hirnsklerose oder das von Hippel-Lindau-Syndrom. In der Mehrzahl der Fälle ist keine prädisponierende Erkrankung zu eruieren.
Die Diagnostik besteht aus einer klinischen Untersuchung und nachfolgend aus einer bildgebenden Untersuchung des Kopfes. Aufgrund ihrer sehr guten Weichteilauflösung ist die Magnetresonanztomographie (MRT) die Methode der Wahl. In seltenen Fällen sind zusätzliche Untersuchungen (Computertomographie (CT), MRT der Wirbelsäule, Augenarzt, HNO-Arzt, Liquor, Angiographie, Blutuntersuchungen, elektrophysiologische Tests) erforderlich.
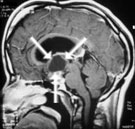
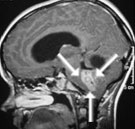
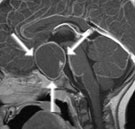
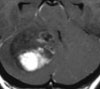
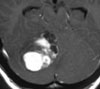
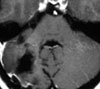
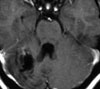
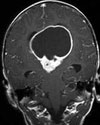
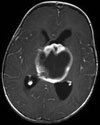
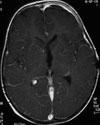
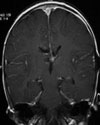
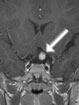
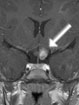
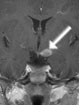
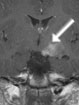
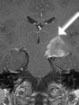
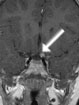
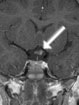
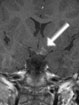
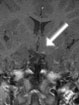
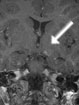
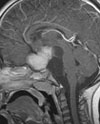
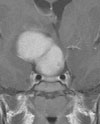
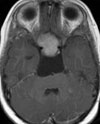
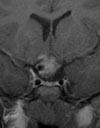
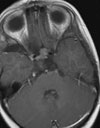
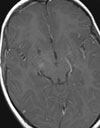
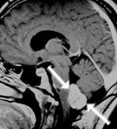
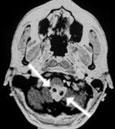
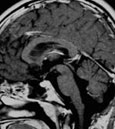
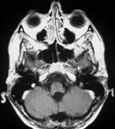
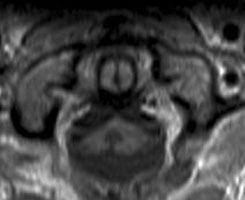
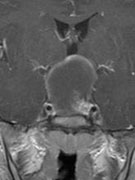
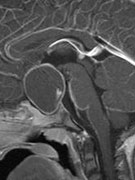
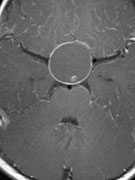
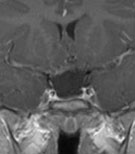
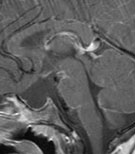
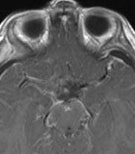
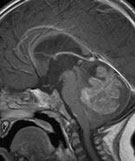
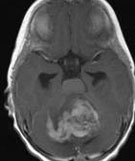
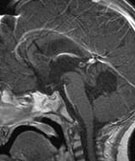
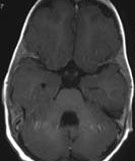
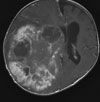
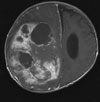
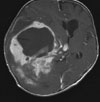
Abb 1: T1-gewichtete MRT-Aufnahmen mit Kontrastmittel von Hirntumoren (Pfeile)
A: Optikusgliom B: Pilozytisches Hirnstammastrozytom C: Kraniopharyngeom
Bei den meisten kindlichen Hirntumoren steht die Operation an erster Stelle der Behandlungskette. Handelt es sich um gutartige Tumoren kann bei vollständiger Tumorentfernung eine Heilung erzielt werden. Ist eine vollständige Tumorentfernung nicht möglich, werden bei gutartigen Tumoren regelmäßige MRT-Kontrollen durchgeführt und bei Wachstumstendenz des Tumorrestes adjuvante Therapien (Bestrahlung und/oder Chemotherapie) durchgeführt. Bei malignen Tumoren erfolgt nach der Operation immer eine Bestrahlung und/oder Chemotherapie, auch wenn der Tumor im MRT postoperativ nicht mehr nachweisbar ist. Eine Bestrahlung sollte jedoch bei Kindern unter 3 Jahren nicht durchgeführt werden, da die Bestrahlung eine erhebliche Langzeittoxizität auf das sich entwickelnde Gehirn hat. In dieser Altersgruppe wird nur eine Chemotherapie durchgeführt. Bei einigen hochmalignen Tumoren kann eine Chemotherapie vor der Operation sinnvoll sein. Bei inoperablen Tumoren, bei denen keine Tumorentfernung durchgeführt werden kann, wird eine Probeentnahme durchgeführt, um die Art des Tumors bestimmen zu können und die entsprechende Therapie einzuleiten.
Pilozytische Astrozytome sind gutartige, langsam wachsende, vom Hirn sehr gut abgegrenzte Tumoren, die häufig im Kleinhirn vorkommen. Im MRT stellen sie sich meist als zystische, stark Kontrastmittel-aufnehmende Läsionen dar. Die Operation ist die Therapie der Wahl. Meist ist eine vollständige Tumorentfernung möglich und die Patienten sind geheilt. Rezidive sind sehr selten.
Abb 2.
A und B: MRT eines 16jährigen Mädchens mit einem pilozytischen Astrozytom des Kleinhirns. Die Patientin klagte über Kopfschmerzen und Gleichgewichtsstörungen.
C und D: Das postoperative MRT zeigt die vollständige Tumorentfernung 2 Jahre nach der Operation. Die Patientin ist beschwerdefrei, keine neurologischen Defizite.
Abb 3.
A und B: MRT eines 4jährigen Mädchens mit einem pilozytischen Astrozytom der Mittellinie (Thalamus ? 3. Hirnkammer). Die Patientin war durch Fallneigung und Gleichgewichtsstörungen auffällig geworden.
C und D: Das postoperative MRT zeigt die vollständige Tumorentfernung. Bis auf eine dezente Gangunsicherheit bestehen keine neurologischen Defizite.
Abb 4.
A und B: MRT eines 14jährigen Jungen mit einem pilozytischen Astrozytom des rechten Schläfenlappens. Der Patient klagte über Kopfschmerzen, Übelkeit und Erbrechen.
C und D: Das postoperative MRT zeigt die vollständige Tumorentfernung 4 Jahre nach der Operation. Der Patient ist beschwerdefrei, keine neurologischen Defizite.
Das Optikusgliom ist eine Sonderform der pilozytischen Astrozytome. Optikusgliome breiten sich entlang der Sehnerven bzw. Sehbahn aus, involvieren jedoch auch häufig das Zwischenhirn (Hypothalamus). Sehstörungen oder hormonelle Störungen sind die häufigsten Symptome. Die Therapieempfehlungen sind sehr widersprüchlich, da der Spontanverlauf der Erkrankung nicht vorhersehbar ist. Spontane Remissionen sind möglich (siehe Abb. 5). Bei raumfordernden Tumoren ist eine Teilresektion sinnvoll. Eine vollständige Tumorentfernung ist nur selten indiziert, da bei größeren Tumoren das Risiko schwerwiegender hypothalamischer Störungen besteht. Bei Tumoren, die auf die Augenhöhle (Orbita) beschränkt sind, kann eine Durchtrennung des Optikus vor der Sehnervenkreuzung vorgenommen werden, um eine Ausbreitung des Tumors auf die Gegenseite zu verhindern.
Abb 5.
A: MRT eines 2jährigen Mädchens mit einem ausgedehnten Optikusgliom (linker Sehnerv-Sehnervenkreuzung-Sehbahn). Die Patientin war durch eine Fallneigung und eine Bevorzugung der linken Hand auffällig geworden. Aufgrund der Tumorausdehnung erfolgte keine Tumorresektion, sondern nur eine Tumorbiopsie.
B: Das Kontroll-MRT nach 6 Monaten zeigt die erstaunliche spontane Rückbildung des Tumors (Tumorgröße seit 2 Jahren stabil).
Abb 6.
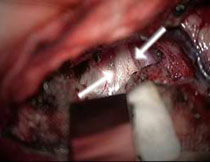
A: MRT eines 3jährigen Jungen mit einem teilweise zystischen Optikusgliom, das den Hirnstamm komprimiert. Der Junge war durch eine Entzündung der Augenhöhle (Orbitaphlegmone) auffällig geworden, und der Tumor als Zufallsbefund diagnostiziert.
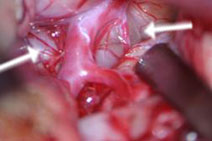
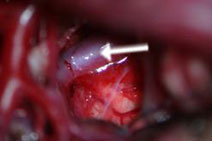
B: Tumor unterhalb der Hirnschlagader (Pfeile). C: Nach Tumorresektion liegt der Hirnstamm mit der Basilararterie frei (Pfeil).
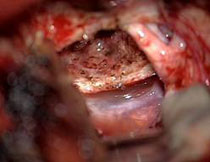
D: Das Kontroll-MRT 3 Monaten nach der Operation zeigt die weitgehende Tumorentfernung. Der Tumoranteil, der den Hypothalamus involviert, wurde belassen, um Zwischenhirnstörungen zu vermeiden. Der kleine Patient ist neurologisch und endokrinologisch unauffällig.
Ependymome sind Tumoren, die von der Auskleidung der Hirnkammern oder des Zentralkanales im Rückenmark (Ependym) ausgehen. Aufgrund ihrer Lage in den Hirnkammern verursachen sie häufig einen Aufstau des Nervenwassers. Die Operation ist die Therapie der Wahl. Ependymome lassen sich meist sehr gut vom normalen Hirngewebe abgrenzen, was für die operative Entfernung sehr hilfreich ist. Nur bei malignen Tumoren oder inoperablen Tumorresten wird eine Bestrahlung durchgeführt.
Abb 7.
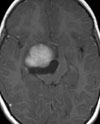
A und B: MRT eines 18jährigen Jungen mit einem Ependymom der Rautengrube (Pfeile). Der Junge war durch Kopfschmerzen, Übelkeit und Erbrechen auffällig geworden.
C: Tumor (Pfeile) unterhalb der Kleinhirntonsillen.
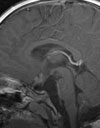
D und E: Das Kontroll-MRT 4 Jahre nach der Operation zeigt die vollständige Tumorentfernung ohne Rezidivhinweis. Der Patient ist neurologisch unauffällig.
Abb 8.
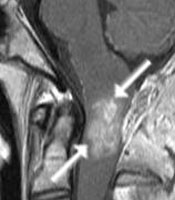
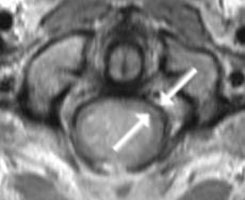
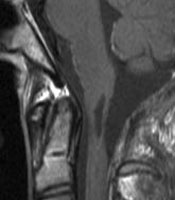
A: MRT eines 16jährigen Jungen mit einem Ependymom des oberen Halsmarkes (Pfeile). Der Junge war durch Nackenschmerzen und Erbrechen auffällig geworden. Trotz der ungünstigen Lage (Gefahr des hohen Querschnittsyndroms mit Atemlähmung sowie Arm- und Beinlähmung) ist die Operation die Therapie der Wahl.
B: Die Pfeile markieren das um den Tumor verbliebene dünne normale Rückenmarkgewebe (grauer Saum um den hellen Tumor herum)
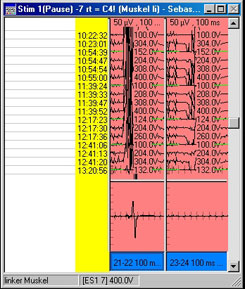
C: Monitoring der motorisch-evozierten Potentiale.
D und E: Das Kontroll-MRT 3 Monate nach der Operation zeigt die vollständige Tumorentfernung. Der Patient ist bis auf eine leichte Ungeschicklichkeit der rechten Hand neurologisch unauffällig.
Kraniopharyngeome sind gutartige Missbildungstumoren, die aus Resten der Rathke?schen Tasche entstehen und meist am Hypophysenstiel ihren Ursprung haben. Sie wachsen langsam und werden durch Kompression der nervalen Strukturen symptomatisch. Bei Ausbreitung bis in die 3. Hirnkammer können sie zu einem Nervenwasseraufstau mit Ausbildung eines Wasserkopfes (Hydrozephalus) führen. Kraniopharyngeome verursachen häufig Sehstörungen durch Kompression der Sehnerven. Bei Nichtbehandlung und Tumorprogredienz droht die Erblindung. Weiterhin werden die Tumoren durch Störungen der hormonellen Regulation auffällig.
Abb 9.
A: MRT eines 13jährigen Mädchens mit einem großen zystischen Kraniopharyngeom. Das Mädchen war durch eine hochgradige Sehminderung und Gesichtsfeldeinschränkung auffällig geworden.
B: Der Tumor hat die Sehnerven stark komprimiert und abgeplattet (Der Pfeil markiert den rechten Sehnerven auf der Tumorkapsel).
C: Operationsfeld nach kompletter Tumorentfernung.
D: Das postoperative Kontroll-MRT zeigt die vollständige Tumorentfernung. Das Sehvermögen konnte erhalten werden.
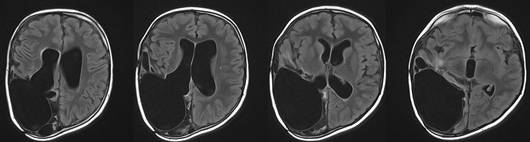
Medulloblastome sind maligne Tumoren (WHO Grad 4), die meist vom Kleinhirnwurm ausgehen. Diese Tumoren neigen zu Absiedlungen in den Nervenwasserraum. Die Operation wird immer ergänzt durch eine postoperative Strahlen- und / oder Chemotherapie.
Abb 10.
A: MRT eines 6jährigen Jungen mit einem großen Medulloblastom des Kleinhirnwurmes. Der Junge wurde durch Erbrechen auffällig.
B: Das postoperative Kontroll-MRT 8 Monate nach der Operation zeigt die vollständige Tumorentfernung. Bis auf leichte Gleichgewichtsstörungen und eine Gangunsicherheit ist der kleine Patient neurologisch unauffällig.
Gangliogliome sind niedrig maligne Mischtumoren (meist WHO Grad 2), die eine gliale und neurale Komponente haben. Sie wachsen langsam und lassen sich sehr gut vom normalen Hirngewebe abgrenzen. Die Operation ist die Therapie der Wahl. Nur bei malignen Tumoren (Grad 3 und 4) erfolgt eine Bestrahlung und / oder Chemotherapie.
Video 1 (2.4 MB)
Zum Starten der Videosequenz bitte auf das Bild klicken.
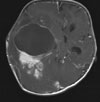
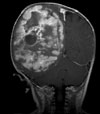
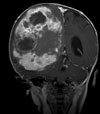
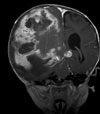
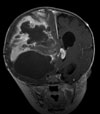
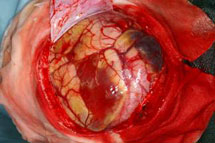
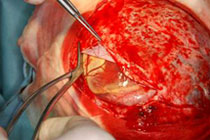
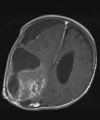
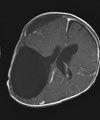
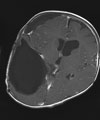
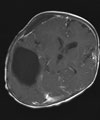
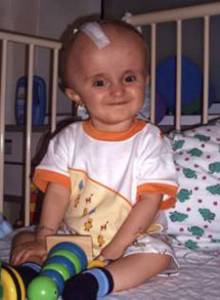
Abb 11.
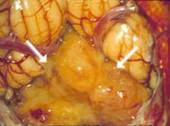
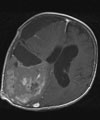
A: MRT eines 1,5jährigen Jungen mit einem monströsen Gangliogliom der rechten Hirnhälfte. Der Junge war durch die vom Tumor verursachte Schädelasymmetrie auffällig geworden. Der Junge erhielt zunächst eine Chemotherapie, die jedoch nicht wirksam war. Der Tumor wuchs weiter. Trotz der enormen Tumorgröße und der sehr guten Durchblutung des Tumors ist die Operation die Therapie der Wahl.
B: Intraoperatives Bild: Der Tumor hat die Hirnrinde bereits durchbrochen.
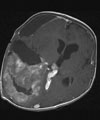
C: Das Kontroll-MRT nach der ersten Operation zeigt die Teilentfernung des Tumors. Aufgrund der starken Durchblutung des Tumors wurde zunächst nur die Hälfte des Tumors entfernt, um einen übermäßigen Blutverlust zu vermeiden.
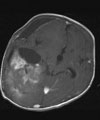
D: Nach 3 Monaten wurde der Resttumor vollständig entfernt. Das Kontroll-MRT 4 Monate nach der Operation zeigt die vollständige Tumorentfernung.
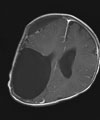
E: Der kleine Patient nach der Operation (links) und im Alter von 12 Jahren (rechts).
Postoperativer Verlauf:
Die Kontroll-MRT-Bildgebung 10 Jahre nach der Operation zeigt weiterhin keinen Rest-Tumor oder ein Tumor-Rezidiv.
Rückfragen an Prof. Dr. med. Henry W. S. Schroeder, Tel.: 03834-86-6162,
Fax: 03834-86-6164, E-Mail: Henry.Schroeder@uni-greifswald.de
|